Pagpapabuti ng energy estimation ng isang chemistry Hamiltonian gamit ang SQD
Sa tutorial na ito ay nag-iimplementa tayo ng isang Qiskit pattern na nagpapakita kung paano i-post-process ang mga noisy quantum samples upang makahanap ng aproksimasyon sa ground state ng isang chemistry Hamiltonian: ang molecule sa equilibrium sa 6-31G basis set. Susundan natin ang isang sample-based quantum diagonalization approach upang i-process ang samples na kinuha mula sa isang 36-qubit quantum circuit ansatz (sa kasong ito, isang LUCJ circuit). Upang isaalang-alang ang epekto ng quantum noise, ginagamit ang configuration recovery technique.
Ang pattern ay maaaring ilarawan sa apat na hakbang:
- Hakbang 1: I-map sa quantum problem
- Bumuo ng ansatz para sa pag-estimate ng ground state
- Hakbang 2: I-optimize ang problem
- I-transpile ang ansatz para sa backend
- Hakbang 3: Isagawa ang mga eksperimento
- Kumuha ng samples mula sa ansatz gamit ang
Samplerprimitive
- Kumuha ng samples mula sa ansatz gamit ang
- Hakbang 4: I-post-process ang resulta
- Self-consistent configuration recovery loop
- I-post-process ang buong set ng bitstring samples, gamit ang prior knowledge ng particle number at ang average orbital occupancy na kinakalkula sa pinakahuling iteration.
- Probabilistically gumawa ng batches ng subsamples mula sa recovered bitstrings.
- I-project at i-diagonalize ang molecular Hamiltonian sa bawat sampled subspace.
- I-save ang minimum ground state energy na natagpuan sa lahat ng batches at i-update ang avg orbital occupancy.
- Self-consistent configuration recovery loop
Para sa halimbawang ito, ang interacting-electron Hamiltonian ay tumatagal ng generic form:
Ang / ay ang fermionic creation/annihilation operators na nauugnay sa -th basis set element at sa spin . Ang at ay ang one- at two-body electronic integrals.
Ang SQD workflow na may self-consistent configuration recovery ay inilalarawan sa sumusunod na diagram.
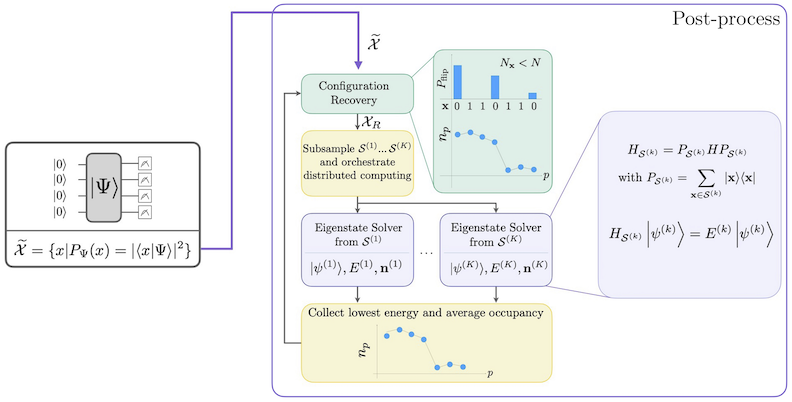
Ang SQD ay kilalang gumagana nang maayos kapag ang target eigenstate ay sparse: ang wave function ay sinusuportahan sa isang set ng basis states na ang laki ay hindi tumataas nang exponential sa laki ng problema. Sa scenario na ito, ang diagonalization ng Hamiltonian na nai-project sa subspace na tinukoy ng :
ay nagbibigay ng magandang aproksimasyon sa target eigenstate. Ang papel ng quantum device ay gumawa ng samples ng mga miyembro ng lamang. Una, ang quantum circuit ay naghahanda ng state sa quantum device. Ginagamit ang Jordan-Wigner encoding. Dahil dito, ang mga miyembro ng computational basis ay kumakatawan sa Fock states (electronic configurations/determinants). Ang circuit ay sinasample sa computational basis, na nagbibigay ng set ng noisy configurations . Ang mga configuration ay kinakatawan ng bitstrings. Ang set ay pagkatapos ay ipinapasa sa classical post-processing block, kung saan ginagamit ang self-consistent configuration recovery technique. Sa SQD framework, ang papel ng quantum device ay gumawa ng probability distribution.
Hakbang 1: I-map ang problem sa isang quantum circuit
Sa tutorial na ito, mag-aaproksima tayo ng ground state energy ng isang molecule. Una, tutukuyin natin ang molecule at ang mga katangian nito. Sunod, gagawa tayo ng local unitary cluster Jastrow (LUCJ) ansatz (quantum circuit) upang bumuo ng samples mula sa isang quantum computer para sa ground state energy estimation.
Una, tutukuyin natin ang molecule at ang mga katangian nito.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import warnings
warnings.filterwarnings("ignore")
import pyscf
import pyscf.cc
import pyscf.mcscf
# Specify molecule properties
open_shell = False
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="6-31g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
num_orbitals = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
num_elec_a = (n_electrons + mol.spin) // 2
num_elec_b = (n_electrons - mol.spin) // 2
cas = pyscf.mcscf.CASCI(scf, num_orbitals, (num_elec_a, num_elec_b))
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), num_orbitals)
# Compute exact energy
exact_energy = cas.run().e_tot
converged SCF energy = -108.835236570775
CASCI E = -109.046671778080 E(CI) = -32.8155692383188 S^2 = 0.0000000
Sunod, gagawa tayo ng ansatz. Ang LUCJ ansatz ay isang parameterized quantum circuit, at i-iinitialize natin ito gamit ang t2 at t1 amplitudes na nakuha mula sa CCSD calculation.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -109.0398256929734 E_corr = -0.2045891221988311
Gagamitin natin ang ffsim package upang gumawa at i-initialize ang ansatz gamit ang t2 at t1 amplitudes na kinakalkula sa itaas. Dahil ang ating molecule ay may closed-shell Hartree-Fock state, gagamitin natin ang spin-balanced variant ng UCJ ansatz, UCJOpSpinBalanced.
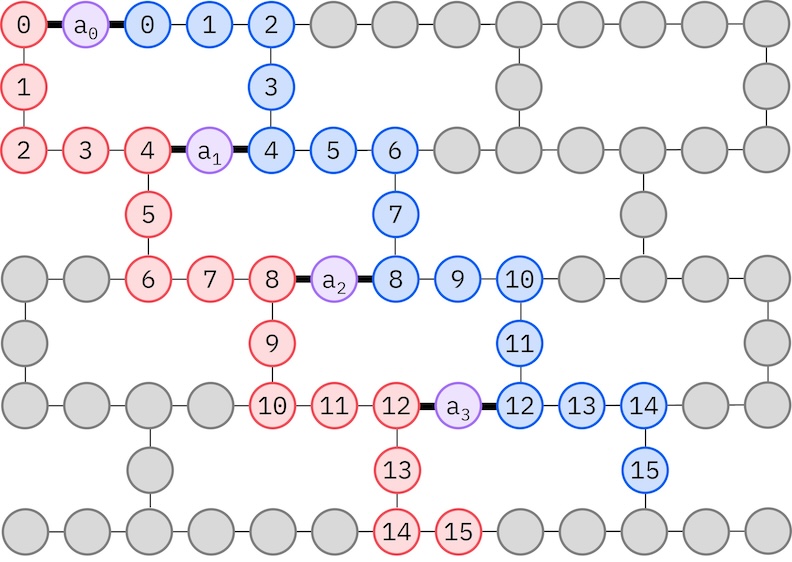
Dahil ang ating target IBM hardware ay may heavy-hex topology, kukuha tayo ng zig-zag pattern para sa qubit interactions. Sa pattern na ito, ang orbitals (kinakatawan ng qubits) na may parehong spin ay konektado gamit ang line topology (red at blue circles) kung saan ang bawat linya ay tumatagal ng zig-zag shape dahil sa heavy-hex connectivity ng target hardware. Muli, dahil sa heavy-hex topology, ang orbitals para sa iba't ibang spins ay may koneksyon sa pagitan ng bawat ika-4 na orbital (0, 4, 8, atbp.) (purple circles).
import ffsim
from qiskit import QuantumCircuit, QuantumRegister
n_reps = 1
alpha_alpha_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
alpha_beta_indices = [(p, p) for p in range(0, num_orbitals, 4)]
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(alpha_alpha_indices, alpha_beta_indices),
)
nelec = (num_elec_a, num_elec_b)
# create an empty quantum circuit
qubits = QuantumRegister(2 * num_orbitals, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(num_orbitals, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Hakbang 2: I-optimize ang problem
Susunod, i-ooptimize natin ang ating circuit para sa target hardware. Kailangan nating pumili ng hardware device na gagamitin bago i-optimize ang ating circuit. Gagamit tayo ng fake 127-qubit backend mula sa qiskit_ibm_runtime upang gayahin ang isang totoong device. Para tumakbo sa isang totoong QPU, palitan lamang ang fake backend ng totoong backend. Tingnan ang Qiskit IBM Runtime docs para sa karagdagang impormasyon.
from qiskit_ibm_runtime.fake_provider import FakeSherbrooke
backend = FakeSherbrooke()
Susunod, inirerekomenda namin ang sumusunod na mga hakbang upang i-optimize ang ansatz at gawin itong hardware-compatible.
- Pumili ng physical qubits (
initial_layout) mula sa target hardware na sumusunod sa zig-zag pattern na inilarawan sa itaas. Ang paglalagay ng qubits sa pattern na ito ay humahantong sa isang efficient hardware-compatible circuit na may mas kaunting gates. - Bumuo ng staged pass manager gamit ang generate_preset_pass_manager function mula sa Qiskit gamit ang iyong pinili na
backendatinitial_layout. - Itakda ang
pre_initstage ng iyong staged pass manager saffsim.qiskit.PRE_INIT. Angffsim.qiskit.PRE_INITay may kasamang Qiskit transpiler passes na nagde-decompose ng gates sa orbital rotations at pagkatapos ay pinagsasama ang orbital rotations, na nagreresulta sa mas kaunting gates sa pinal na circuit. - Patakbuhin ang pass manager sa iyong circuit.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
spin_a_layout = [0, 14, 18, 19, 20, 33, 39, 40, 41, 53, 60, 61, 62, 72, 81, 82]
spin_b_layout = [2, 3, 4, 15, 22, 23, 24, 34, 43, 44, 45, 54, 64, 65, 66, 73]
initial_layout = spin_a_layout + spin_b_layout
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend, initial_layout=initial_layout
)
# without PRE_INIT passes
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/o pre-init passes): {isa_circuit.count_ops()}")
# with PRE_INIT passes
# We will use the circuit generated by this pass manager for hardware execution
pass_manager.pre_init = ffsim.qiskit.PRE_INIT
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/ pre-init passes): {isa_circuit.count_ops()}")
Gate counts (w/o pre-init passes): OrderedDict({'rz': 4420, 'sx': 3432, 'ecr': 1366, 'x': 239, 'measure': 32, 'barrier': 1})
Gate counts (w/ pre-init passes): OrderedDict({'rz': 2460, 'sx': 2156, 'ecr': 730, 'x': 71, 'measure': 32, 'barrier': 1})
Hakbang 3: Isagawa ang mga eksperimento
Pagkatapos i-optimize ang circuit para sa hardware execution, handa na tayong patakbuhin ito sa target hardware at mangolekta ng samples para sa ground state energy estimation. Dahil isa lamang circuit ang mayroon tayo, gagamitin natin ang Job execution mode ng Qiskit Runtime at ie-execute natin ang ating circuit.
Tandaan: Na-comment out namin ang code para sa pagpapatakbo ng circuit sa isang QPU at iniwan ito para sa reference ng user. Sa halip na tumakbo sa totoong hardware sa guide na ito, gagawa lamang tayo ng random samples na kinuha mula sa uniform distribution.
import numpy as np
from qiskit_addon_sqd.counts import generate_bit_array_uniform
# from qiskit_ibm_runtime import SamplerV2 as Sampler
# sampler = Sampler(mode=backend)
# job = sampler.run([isa_circuit], shots=10_000)
# primitive_result = job.result()
# pub_result = primitive_result[0]
# bit_array = pub_result.data.meas
rng = np.random.default_rng(24)
bit_array = generate_bit_array_uniform(10_000, num_orbitals * 2, rand_seed=rng)
Hakbang 4: I-post-process ang resulta
Ngayon, pinapatakbo natin ang SQD algorithm gamit ang diagonalize_fermionic_hamiltonian function. Tingnan ang API documentation para sa mga paliwanag ng arguments sa function na ito.
Ang solver na kasama sa SQD addon ay gumagamit ng implementation ng PySCF ng selected CI, partikular na ang pyscf.fci.selected_ci.kernel_fixed_space. Ang halimbawa sa ibaba ay nagpapakita rin kung paano magpasa ng keyword arguments sa function na iyon sa pamamagitan ng kasamang solver. Dito ipinapasa natin ang max_cycle argument.
from functools import partial
from qiskit_addon_sqd.fermion import SCIResult, diagonalize_fermionic_hamiltonian, solve_sci_batch
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 1
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}")
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=num_orbitals,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -105.45358671756313
Subspace dimension: 5476
Iteration 2
Subsample 0
Energy: -107.95172900082163
Subspace dimension: 249001
Iteration 3
Subsample 0
Energy: -108.97460330369815
Subspace dimension: 339889
Iteration 4
Subsample 0
Energy: -109.02739376648793
Subspace dimension: 440896
Iteration 5
Subsample 0
Energy: -109.030972328451
Subspace dimension: 597529
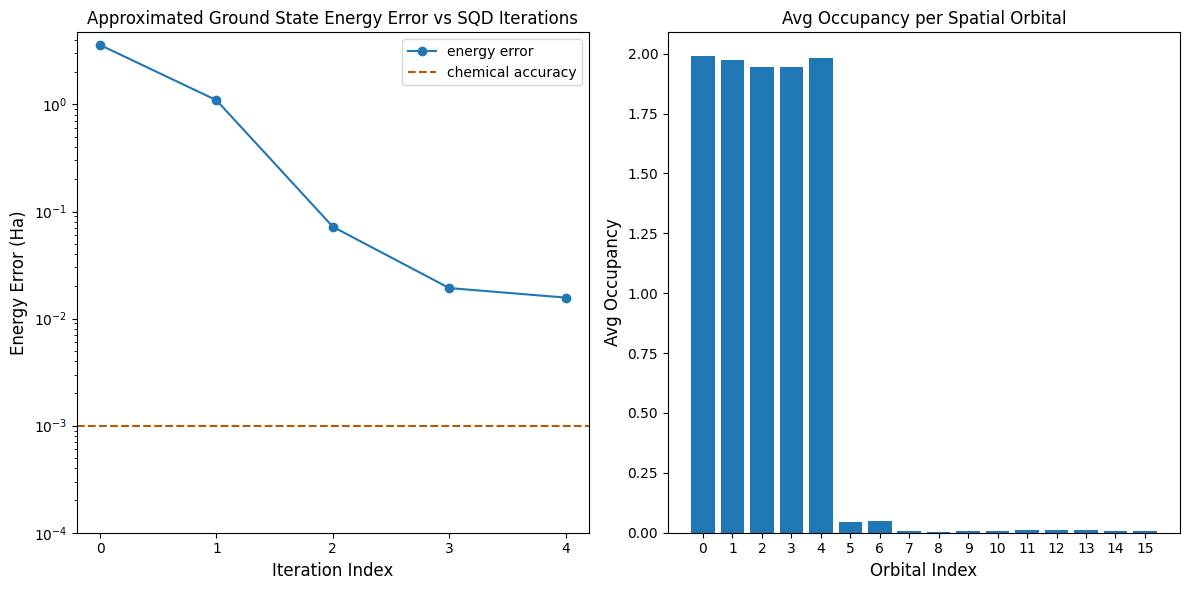
Ngayon, ipla-plot natin ang mga resulta.
Ang unang plot ay nagpapakita na pagkatapos ng ilang iterations, ina-estimate natin ang ground state energy sa loob ng ~16 mH (ang chemical accuracy ay karaniwang tinatanggap na 1 kcal/mol 1.6 mH). Tandaan, ang quantum samples sa demo na ito ay puro noise. Ang signal dito ay nagmumula sa a priori knowledge ng electronic structure at molecular Hamiltonian.
Ang pangalawang plot ay nagpapakita ng average occupancy ng bawat spatial orbital pagkatapos ng pinal na iteration. Makikita natin na ang spin-up at spin-down electrons ay parehong sumasakop sa unang limang orbitals na may mataas na probability sa ating mga solusyon.
import matplotlib.pyplot as plt
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - exact_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(y=chem_accuracy, color="#BF5700", linestyle="--", label="chemical accuracy")
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Exact energy: {exact_energy:.5f} Ha")
print(f"SQD energy: {min_e[-1]:.5f} Ha")
print(f"Absolute error: {e_diff[-1]:.5f} Ha")
plt.tight_layout()
plt.show()
Exact energy: -109.04667 Ha
SQD energy: -109.03097 Ha
Absolute error: 0.01570 Ha