HI-VQE Chemistry - Isang Qiskit Function ng Qunova Computing
# Added by doQumentation — required packages for this notebook
!pip install -q qiskit-ibm-catalog qiskit-ibm-runtime
# This cell is hidden from users
from qiskit_ibm_runtime import QiskitRuntimeService
service = QiskitRuntimeService()
instance = service.active_account()["instance"]
backend_name = service.least_busy(operational=True, min_num_qubits=16).name
Tingnan ang API reference
Ang mga Qiskit Function ay isang experimental na feature na available lamang sa mga gumagamit ng IBM Quantum® Premium Plan, Flex Plan, at On-Prem (sa pamamagitan ng IBM Quantum Platform API) Plan. Nasa preview release status ang mga ito at maaaring magbago.
Package versions
Ang code sa pahinang ito ay binuo gamit ang mga sumusunod na kinakailangan. Inirerekomenda naming gamitin ang mga bersyong ito o mas bago.
qiskit-ibm-runtime~=0.45.0
Pangkalahatang-ideya
Sa quantum chemistry, ang electronic structure problem ay nakatuon sa paghahanap ng mga solusyon sa electronic Schrödinger equation — ang mga quantum wave function na naglalarawan ng gawi ng mga elektron ng sistema. Ang mga wave function na ito ay mga vector ng mga complex amplitude, kung saan ang bawat amplitude ay kumakatawan sa kontribusyon ng isang posibleng electron configuration.
Ang ground state ay ang pinakamababang energy wave function ng sistema at may espesyal na kahalagahan sa pag-aaral ng mga molecular system. Ang pinaka-tumpak na paraan para kalkulahin ang ground state ay isinasaalang-alang ang lahat ng posibleng electron configuration, ngunit ito ay nagiging imposible para sa mas malalaking sistema dahil ang bilang ng mga configuration ay lumalaki nang eksponensyal habang lumalaki ang sistema.
Ang Handover Iterative Variational Quantum Eigensolver (HI-VQE) ay isang makabagong hybrid quantum-classical na pamamaraan para sa tumpak na pagtatantya ng ground state ng mga molecular system. Pinagsasama nito ang quantum hardware at classical computing, gumagamit ng mga quantum processor para mahusay na tuklasin ang mga kandidatong electron configuration at kinakalkula ang nagresultang wave function sa mga classical computer. Sa pamamagitan ng pagbuo ng mga compact ngunit chemically accurate na wave function, pinapabuti ng HI-VQE ang pananaliksik at pagtuklas sa quantum chemistry at materials science.

Binabawasan ng HI-VQE ang computational complexity ng electronic structure problem sa pamamagitan ng mahusay na pagtatantya ng ground state nang may mataas na katumpakan. Nakatuon ito sa isang maingat na piniling subset ng mga pinaka-kaugnay na electron configuration, ino-optimize ang parehong katumpakan at kahusayan.
Sa pagsasama ng mga kalakasan ng parehong classical at quantum computer, paulit-ulit na pinipino at pinapabuti ng HI-VQE ang kasalukuyang tinatantyang wave function. Ang natatanging mga teknik sa pagbuo ng subspace nito ay tumutulong na gawing mas mahusay ang pagpili ng configuration, para ang mga gumagamit ay may mas malaking computational control at pinahusay na katumpakan sa mga quantum chemistry simulation.
Kung gusto mong matuto nang mas malalim tungkol sa algorithm, maaari kang basahin ang kaugnay na research paper.
Paglalarawan
Ang bilang ng mga electron configuration para sa isang molecular system ay lumalaki nang eksponensyal habang lumalaki ang sistema. Gayunpaman, para sa ilang partikular na electronic state, tulad ng ground state, karaniwan na isang maliit na bahagi lamang ng mga configuration ang may malaking kontribusyon sa energy ng estado. Sinasamantala ng mga Selected Configuration Interaction (SCI) na pamamaraan ang paguusok na ito para mabawasan ang mga gastos sa computation sa pamamagitan ng pagtukoy at pagtuon sa mga pinaka-kaugnay na configuration. Ang subset ng mga configuration na ito ay tinatawag na subspace.
Ginagamit ng HI-VQE ang likas na kahusayan ng mga quantum computer sa pagrepresenta ng mga molecular system para tulungan ang paghahanap ng subspace. Pinagsasama nito ang mga classical at quantum subroutine para malutas ang electronic structure problem nang may mataas na katumpakan. Hindi tulad ng mga umiiral na quantum SCI na pamamaraan, pinagsasama ng HI-VQE ang variational training, iterative subspace construction, at pre-diagonalization configuration screening para mapahusay ang kahusayan sa pamamagitan ng pagbabawas ng mga quantum measurement, iteration, at classical diagonalization cost. Kaya naman, maaaring ilapat ang HI-VQE sa mas malalaking molecular system na nangangailangan ng mas maraming qubit, at binabawasan ang gastos para malutas ang isang problemang may partikular na laki sa parehong antas ng katumpakan.
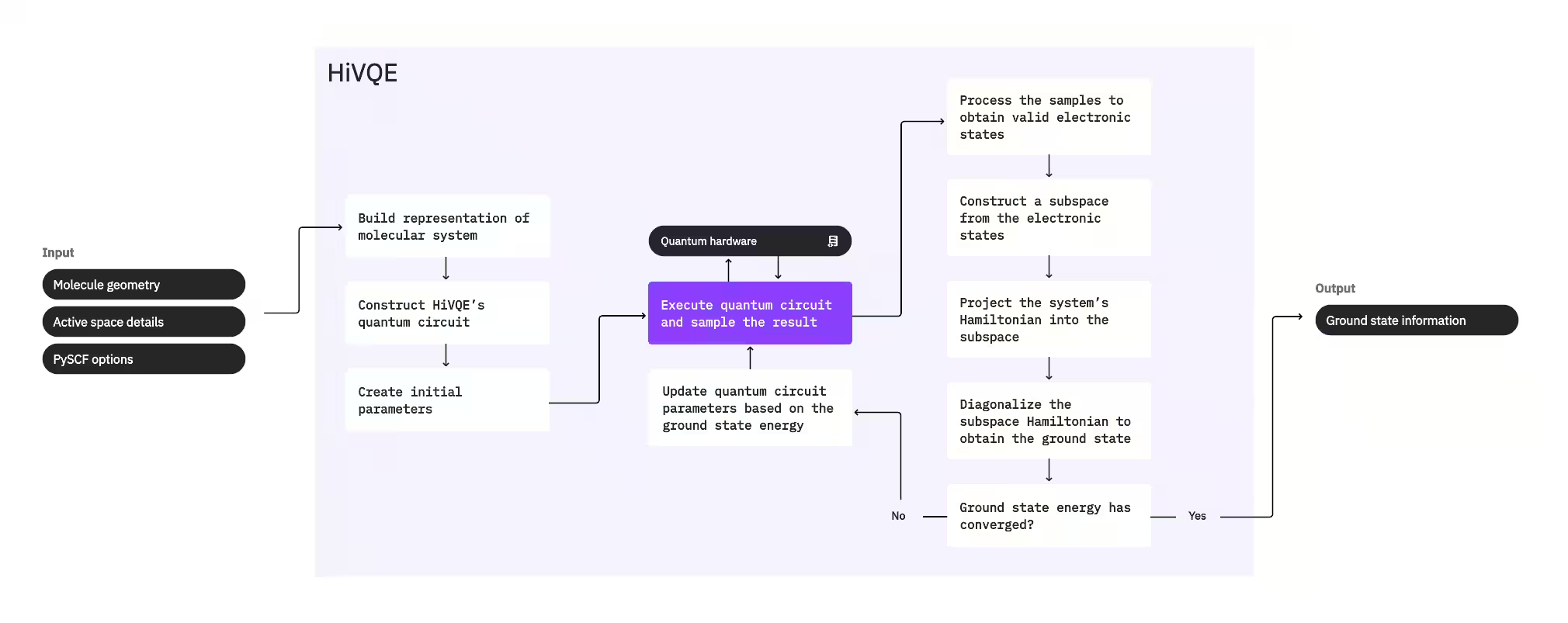
Para kalkulahin ang ground state ng isang sistema, unang ginagamit ng HI-VQE ang classical chemistry package na PySCF para bumuo ng molecular representation mula sa mga input na ibinigay ng gumagamit, tulad ng molecular geometry at iba pang impormasyon tungkol sa molekula. Pagkatapos ay pumapasok ito sa isang hybrid quantum-classical optimization loop, paulit-ulit na pinipino ang isang subspace para optimal na marepresenta ang ground state habang pinipigilan ang bilang ng mga kasama sa configuration. Nagpapatuloy ang loop hanggang matugunan ang mga convergence criteria, tulad ng subspace size o energy stability, pagkatapos ay ini-output ang computed ground state wave function at energy. Maaaring gamitin ang mga resultang ito para bumuo ng tumpak na potential energy surface at magsagawa ng karagdagang pagsusuri ng sistema.
Ang optimization loop ay nakatuon sa pag-aayos ng mga parameter ng quantum circuit para makabuo ng mataas na kalidad na subspace. Nag-aalok ang HI-VQE ng tatlong opsyon sa quantum circuit: excitation_preserving, efficient_su2, at LUCJ. Ang optimization ay nagsisimula malapit sa Hartree-Fock reference state dahil sa pangkalahatang angkop nito. Ang circuit ay isinasagawa pagkatapos sa isang quantum device at ang mga configuration ay sinasample mula sa nagresultang quantum state bago ibalik bilang binary string. Dahil sa ingay ng quantum device, ang ilang sampled na configuration ay maaaring hindi pisikal na wasto, nabigo ang pag-conserve ng electron number o spin. Tinutugunan ito ng HI-VQE gamit ang configuration recovery process mula sa package na qiskit-addon-sqd, para ang mga gumagamit ay makakapag-correct ng mga hindi wastong configuration o itatapon ang mga ito.
Ang mga wastong configuration ay dumadaan sa isang opsyonal na screening step para alisin ang mga inaasahang minimal ang kontribusyon. Binabawasan nito ang dimensyon ng subspace, kaya naman mas mababang gastos ang diagonalization step. Kung pinagana ang screening, isang paunang subspace Hamiltonian ang binubuo mula sa mga wastong configuration at isang diagonalization ang isinasagawa na may maluwag na termination criteria. Kahit mababa ang katumpakan ng mga nagresultang amplitude para sa bawat configuration, ito ay epektibo para hulaan kung aling mga configuration ang dapat iwanan sa labas ng subspace sa iteration na ito, at mabilis itong kalkulahin.
Ang mga piniling configuration ay idadagdag sa subspace, at ang Hamiltonian ng sistema ay ini-project sa subspace na ito. Ang subspace ay paulit-ulit na naa-update, pinapanatili ang mga pinaka-kaugnay na configuration sa buong mga iteration. Ang diskarteng ito ay kaibahan sa mga alternatibong pamamaraan dahil ang quantum circuit ay hindi kailangang tantiyahin ang buong ground state sa bawat hakbang.
Susunod, ang subspace Hamiltonian ay classically diagonalized para makuha ang pinakamababang eigenvalue at ang kaukulang eigenvector nito, na kumakatawan sa isang approximation ng ground state at ng energy nito. Habang nag-iimprove ang kalidad ng subspace sa pamamagitan ng mga iteration, mas malapitan ng computed ground state ang tunay na ground state. Isang karagdagang screening step ay maaaring isagawa sa puntong ito para alisin ang anumang configuration mula sa subspace na walang malaking kontribusyon sa computed ground state. Tinitiyak ng hakbang na ito na ang subspace na dadalhin sa susunod na iteration ay kasing-compact ng posible. Sinusuri ito batay sa mga amplitude na ibinalik ng diagonalization, dahil kinakatawan ng mga ito ang kontribusyon ng bawat configuration sa computed ground state.
Pagkatapos, tinutukoy ng convergence check kung ang karagdagang training ay magpapabuti ng mga resulta. Kung oo, isang opsyonal na classical expansion step ang isasagawa, ia-update ang mga parameter ng quantum circuit para higit pang mabawasan ang computed energy, at uulitin ang proseso. Ang classical expansion step ay nagbibigay ng karagdagang mga configuration para sa subspace, dinadagdagan ang mga configuration na sinample mula sa quantum device. Una nitong tinutukoy ang configuration na may pinakamalaking amplitude sa mga resulta ng diagonalization, bago bumuo ng mga bagong configuration na may single at double excitation mula sa natukoy na configuration. Ang desired na bilang ng mga configuration na ito ay idadagdag sa subspace.
Kapag natukoy na ang mga iteration ay nag-converge na, ibinabalik ng HI-VQE ang computed ground state (sa anyo ng mga estado sa subspace at ang kanilang mga amplitude sa ground state wave function), ang energy nito, at isang energy variance measure na nagbibigay ng indikasyon kung ang computed state ay bumubuo ng isang eigenstate ng Hamiltonian ng sistema.
Maaaring magdesisyon ang mga gumagamit ng quantum circuit na gagamitin at ang bilang ng mga shot na kukunin para sa bawat quantum circuit, pati na rin kontrolin ang subspace size o paganahin ang classical generation ng karagdagang configuration para tulungan ang mga configuration na galing sa quantum. Sa ganitong paraan, maaaring i-customize ng mga gumagamit ang gawi ng HI-VQE ayon sa kanilang nais na mga aplikasyon.
Paglilisensya
Pakitandaan na ang paggamit ng Qiskit Function na ito ay limitado sa mga problemang nangangailangan ng hindi hihigit sa 20 qubit, maliban kung makakuha ng lisensya na nagbibigay ng mas mataas na limitasyon.
Mangyaring mag-email sa qiskit.support@qunovacomputing.com kung nais mong magtanong tungkol sa pagkuha ng lisensya.
Pagsisimula
Una, humiling ng access sa function. Pagkatapos, i-authenticate gamit ang iyong IBM Quantum® API key at, kung nai-save na nito ang iyong account sa iyong lokal na environment, piliin ang Qiskit Function tulad ng sumusunod:
import reprlib
from qiskit_ibm_catalog import QiskitFunctionsCatalog
catalog = QiskitFunctionsCatalog(channel="ibm_quantum_platform")
function = catalog.load("qunova/hivqe-chemistry")
Halimbawa
Ang unang halimbawa ay nagpapakita kung paano kalkulahin ang ground state energy para sa isang NH3 na molekula gamit ang HI-VQE algorithm.
Tukuyin ang molecular geometry at mga opsyon
Ang molecular geometry ng NH3 ay ibinibigay na may mga Cartesian coordinate na pinaghihiwalay ng ";" para sa bawat atom.
# Define the molecule geometry
geometry = """
N -0.85188 -0.02741 0.03141;
H 0.16545 0.00593 -0.01648;
H -1.16348 -0.39357 -0.86702;
H -1.16348 0.94228 0.06281;
"""
Maaaring tukuyin at ibigay ang karagdagang mga opsyon para sa molecular system sa sumusunod na format ng dictionary.
# Configure some options for the job.
molecule_options = {"basis": "sto3g"}
hivqe_options = {"shots": 100, "max_iter": 20}
Isagawa ang function na may geometry at mga input na opsyon.
# Run HI-VQE
job = function.run(
geometry=geometry,
# `backend_name` is the name of a backend with at least 16 qubits,
# for example, "ibm_marrakesh".
backend_name=backend_name,
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
Magandang ideya na i-print ang Function job ID para maibigay ito sa mga support request kung may magkamaling mangyari.
print("Job ID:", job.job_id)
Job ID: e5ced6f2-fd1d-4244-a6aa-bd27cfb0cdee
Ang halimbawang ito ay gumagamit ng 16 qubit na may 8 orbital ng sto3g basis para sa isang NH3 na molekula. Suriin ang status ng iyong Qiskit Function workload o kunin ang mga resulta tulad ng sumusunod:
print(job.status())
QUEUED
Pagkatapos makumpleto ang job, maaaring makuha ang mga resulta gamit ang result() instance.
result = job.result()
# Output can be long, so we display a shortened representation
shortened_result = reprlib.repr(result)
print(shortened_result)
{'eigenvector': [0.9824448589364075, 0.009527106392132133, 6.854074372058527e-08, 3.591500190038039e-07, 0.0012975231577544268, 2.310159709002111e-05, ...], 'energy': -55.52108557170985, 'energy_history': [-55.51901898989887, -55.52056881448526, -55.52065046778772, -55.520690696813716, -55.520691108428, -55.520708448092634, ...], 'energy_variance': 3.066239097617371e-10, ...}
Para ma-access ang ground state energy, gamitin ang "energy" key. Ang "eigenvector" key ay nagbibigay ng mga CI coefficient na may kaukulang bitstring notation ng electron configuration na nakaimbak sa "states" ng mga resulta.
fci_energy = -55.521148034704126 # the exact energy using FCI method
hivqe_energy = result["energy"]
print(
f"|Exact Energy - HI-VQE Energy|: "
f"{abs(fci_energy - hivqe_energy) * 1000} mHa"
)
print(f"Sampled Number of States: {len(result['states'])}")
|Exact Energy - HI-VQE Energy|: 0.06246299427914437 mHa
Sampled Number of States: 1936
Pagganap
Ang seksyong ito ay nagpapakita ng mga ipinahayag na benchmark calculation ng HI-VQE na may 24-qubit na kaso para sa Li2S, isang 40-qubit na kaso para sa isang N2 na molekula, at isang 44-qubit na kaso para sa isang FeP-NO system.
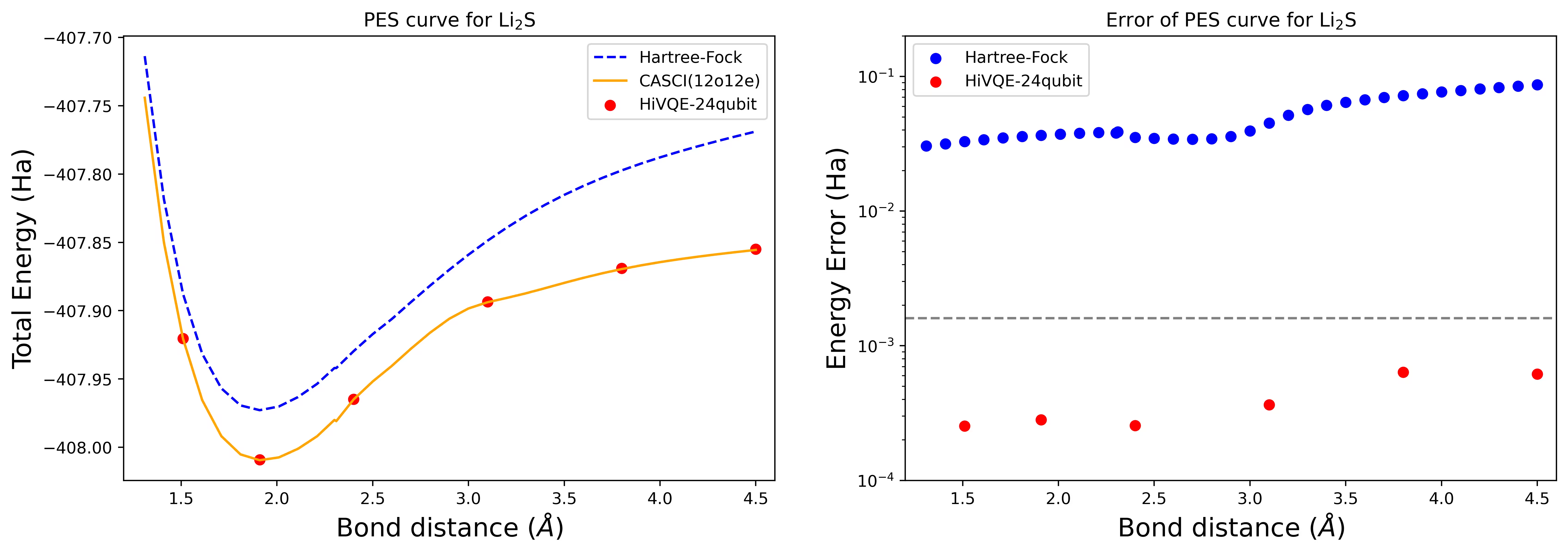
Dissociation potential energy surface curve para sa isang Li2S na molekula na may 24 qubit
Ang PES curve ay ipinapakita kasama ang FCI reference at paunang hula mula sa RHF, kasama ang energy error mula sa FCI reference.
 .
.
Ang mga kalkulasyon ay isinagawa gamit ang mga sumusunod na geometry at opsyon.
# This cell is hidden from users
backend_name = service.least_busy(operational=True, min_num_qubits=38).name
# Define Li2S geometries
Li2S_geoms = {
"Li2S_1.51": "S -1.239044 0.671232 -0.030374;Li -1.506327 0.432403 -1.498949;Li -0.899996 0.973348 1.826768;",
"Li2S_2.40": "S -1.741432 0.680397 0.346702;Li -0.529307 0.488006 -1.729343;Li -1.284307 0.989409 2.177209;",
"Li2S_3.80": "S -2.707255 0.674298 0.909161;Li 0.079218 0.552012 -1.671656;Li -0.927010 0.931502 1.557063;",
}
# Configure some options for the job.
molecule_options = {
"basis": "sto3g",
}
hivqe_options = {
"shots": 100,
"max_iter": 20,
}
results = []
for geom in ["Li2S_1.51", "Li2S_2.40", "Li2S_3.80"]:
# Run HI-VQE
job = function.run(
geometry=Li2S_geoms[geom],
backend_name=backend_name, # can use any device with at least 38 qubits
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
results.append(job.result())
Ang mga pulang tuldok ay kumakatawan sa mga resulta ng HI-VQE calculation para sa anim na iba't ibang geometry, at tatlong geometry na tumutugma sa 1.51, 2.40, at 3.80 Angstrom ang ibinibigay bilang input sa cell sa itaas.
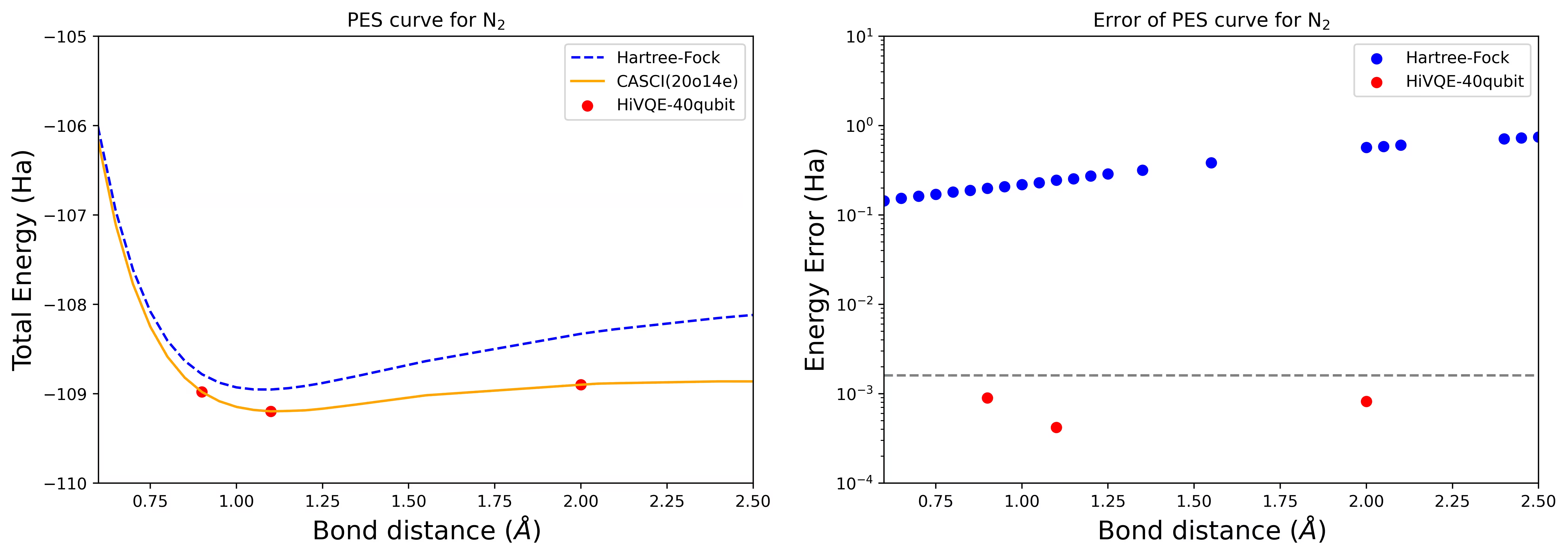
Dissociation PES curve para sa isang N2 na molekula na may 40 qubit
Ang nitrogen na molekula ay natukoy bilang isang multireference system na may malalaking kontribusyon ng correlation energy na higit pa sa Hartree-Fock state. Nagsagawa kami ng benchmark calculation para sa N2 na molekula na may cc-pvdz basis, (20o,14e) gamit ang homo-lumo active orbital selection. Ang bilang ng complete active space (CAS) para irepresenta ang problemang ito ay 6,009,350,400. Hindi posible na makuha ang eigenvalue problem solution (para sa energy at electronic structure) na may ganitong bilang ng mga estado gamit ang isang makapangyarihang desktop computer (16cpu/64GB). Sa HI-VQE, maaaring mahusay na hanapin ng mga gumagamit ang subspace ng mga CAS state para mahanap ang mga chemically accurate na resulta habang nananatiling makatipid ng mga computational resource. Ang mga sumusunod na plot ay nagpapakita ng PES curve ng 40-qubit HI-VQE calculation ng dissociation ng N2 na molekula.
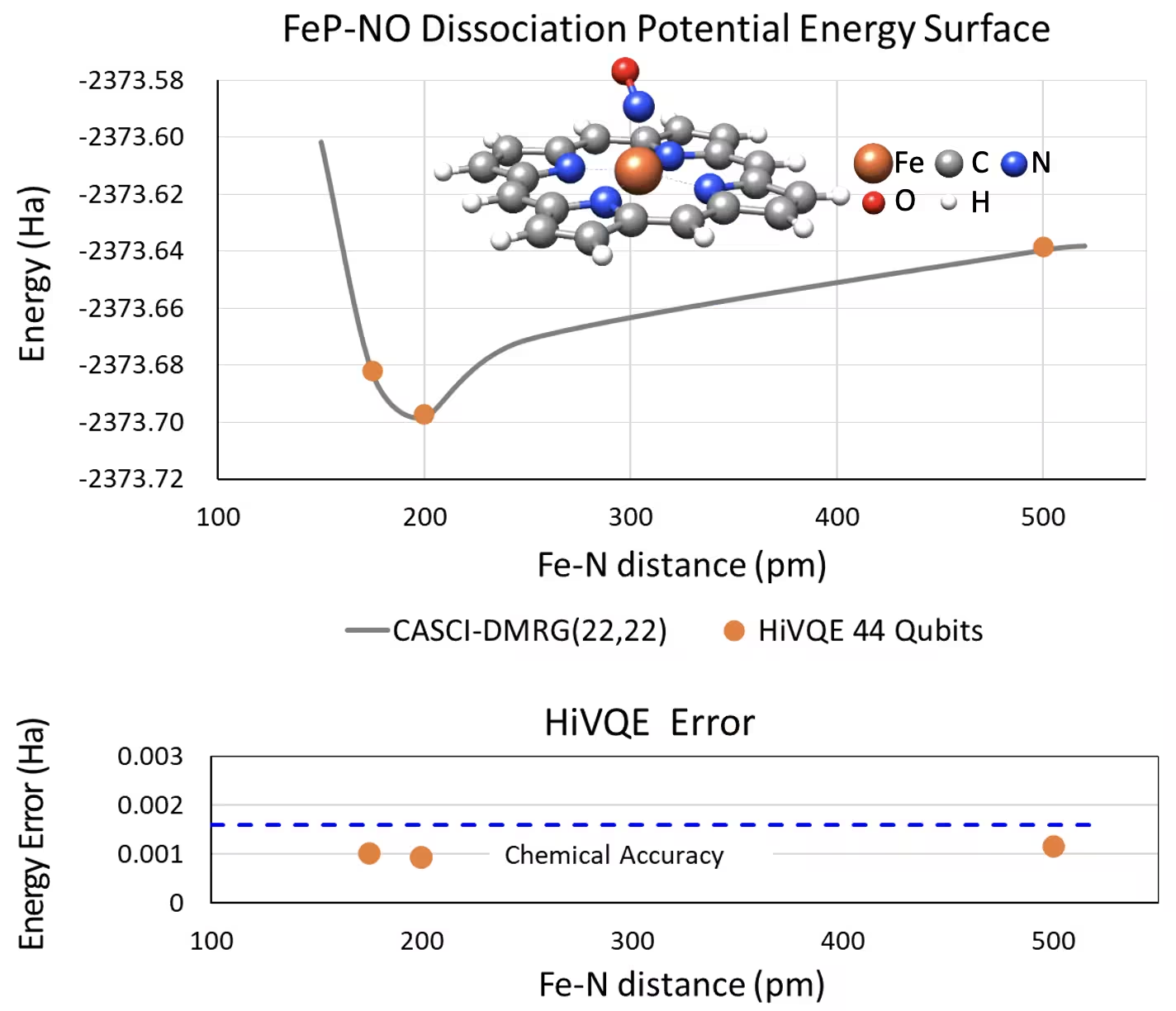
Dissociation PES curve para sa five-coordinated iron(II)-porphyrin na may NO system na may 44 qubit
Isa pang kawili-wiling chemical system ay ang isang iron(II)-porphyrin (FeP) complex na may coordinated nitric oxide (NO) ligand, na kumakatawan sa isang biologically relevant metalloporphyrin system na may mahalagang papel sa iba't ibang physiological process. Sa halimbawang ito, ginamit ang HI-VQE para tantiyahin ang tumpak na potential energy surface curve ng intermolecular interaction sa pagitan ng FeP at NO (ground state energy para sa iba't ibang pinaghiwalay na geometry). Ang pinagsanib na sistema ay may 450 orbital at 202 elektron (450o,202e) na may 6-31g(d) basis sa kabuuan. Ang homo-lumo active orbital selection ay ginamit para kalkulahin ang mas maliit na kaso mula sa tunay na kaso na may (22o,22e). Mula sa mga sumusunod na benchmark result, nakamit namin ang chemical accuracy (> 1.6 mHa) kasama ang state-of-the-art classical computer chemistry calculation ng CASCI(DMRG) (22o,22e) reference.
Mga Benchmark
- Ang exact matrix size ay ang bilang ng mga determinant para sa eksaktong solusyon, tulad ng FCI at CASCI.
- Ang HI-VQE calculation ay nagsa-sample at kinakalkula ang subspace nito (katulad ng, HI-VQE matrix size).
- Ang kabuuang oras ay kinabibilangan ng QPU runtime at Qiskit Function runs na may CPU.
- Ang katumpakan ay tinatantya mula sa energy difference mula sa eksaktong solusyon.
| Chemical system | Bilang ng qubit | Exact matrix size | HI-VQE matrix size | E(diff) mula sa exact (mHa) | Bilang ng iteration | Kabuuang oras | QPU runtime usage |
|---|---|---|---|---|---|---|---|
| (8o,10e) | 16 | 3136 | 1936 | 0.08 | 6 | 37 s | 34 s |
| (10o,10e) | 20 | 63504 | 3969 | 0.60 | 5 | 250 s | 50 s |
| (15o,10e) | 30 | 9018009 | 49729 | 0.90 | 5 | 354 s | 54 s |
| (16o,14e) | 32 | 130873600 | 1798281 | 1.10 | 9 | 6531 s | 121 s |
| (18o,24e) | 36 | 344622096 | 399424 | 0.90 | 24 | 5174 s | 130 s |
| (20o,14e) | 40 | 6009350400 | 9012004 | 1.20 | 21 | 46547 s | 258 s |
Kunin ang mga mensahe ng error
Kung mabigo ang iyong workload, ang status ay magiging ERROR at ang pagtawag sa job.result() ay magtataas ng exception:
job = function.run(
geometry="invalid-geometry", # This will cause an error
backend_name=backend_name,
max_states=2000,
max_expansion_states=15,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
job.result()
job.status()
'ERROR'
Kumuha ng suporta
Maaari kang magpadala ng email sa qiskit.support@qunovacomputing.com para sa tulong sa function na ito.
Kung gusto mong makatulong sa pag-troubleshoot ng isang partikular na error, mangyaring ibigay ang Function job ID ng job na nakaranas ng error.
Mga susunod na hakbang
- Humiling ng access sa function sa pamamagitan ng pagpuno ng form na ito.
- Bisitahin ang API reference para sa Qiskit Function na ito.
- Subukan ang tutorial na Compute dissociation PES curve for FeP-NO with HI-VQE.
- Suriin ang Pellow-Jarman, A., et al. (2025). HIVQE: Handover Iterative Variational Quantum Eigensolver for Efficient Quantum Chemistry Calculations. arXiv preprint arXiv:2503.06292.
- Subukan ang tutorial na Dissociation PES curves with Qunova HiVQE.